Introduction to Fiber Diffraction
Some Macromolecules form Fibers rather than Crystals
Many biological macromolecules will not or cannot crystallize. However, an important group of fibrous macromolecules such as DNA or many of the components of the cytoskeleton form orientated fibers in which the axes of the long polymeric structures are parallel to each other. Often, as in the case of muscle fibers, the orientation is intrinsic; sometimes the long molecules can be induced to form orientated fibers by pulling them from a gel with tweezers, sometimes by flowing a gel through a capillary tube, or even by subjecting them to intense magnetic fields. The experimental set-up is rather simple: the orientated fiber is placed in a collimated x-ray beam at right angles to the beam and the "fiber diffraction pattern" is recorded on a film placed a few cm away from the fibre.
Fibers show helical symmetry rather than the three-dimensional symmetry taken on by crystals. By analysing the diffraction from orientated fibers one can deduce the helical symmetry of the molecule and in favourable cases one can deduce the structure. In general this is done by constructing a model of the fiber (as in DNA) and then calculating the expected diffraction pattern. By comparing the calculated and observed diffraction patterns one eventually arrives at a better model.
Fiber diffraction patterns fall into two main classes: crystalline and non-crystalline.
 the long fibrous molecules are arranged parallel to each other but each molecule takes on a random orientation around the c-axis. The resulting diffraction pattern (right of figure h1) is also based on layer-lines, which reflect the periodic repeat of the fibrous molecule. The intensity along the layer-lines is continuous and can be calculated via a \"Fourier-Bessel Transform\" of the repeating structure of the fibrous molecule (the Fourier-Bessel Transform replaces the Fourier transform used in standard crystallography - The Fourier-Bessel transform arises because of the cylindrical symmetry).")

© Kenneth C. Holmes
Fig h1: Diffraction from the A and B forms of DNA
© Kenneth C. Holmes
In the crystalline case (e.g. A-form of DNA) The long fibrous molecules pack to form long thin micro-crystals which share a common axis (usually referred to as the c-axis). The micro-crystals are randomly arranged around this axis. The resulting diffraction pattern (on left of figure h1) is equvalent to taking one long crystal and spinning it about its axis during the x-ray exposure. All Bragg reflexions are registered at one time. The reflexions are grouped along "layer-lines" which arise from the repeating structure along the c-axis. However, particularly at high resolution the Bragg reflexions tend to fall on top of each other. If the Bragg reflexions could be separated and measured out to high resolution then the standard methods we have described for crystals could be used. Unfortunately this is never the case and model building must be employed - as is generally the case for non-crystalline fibers.
In non-crystalline fibers (e.g. B-form of DNA) the long fibrous molecules are arranged parallel to each other but each molecule takes on a random orientation around the c-axis. The resulting diffraction pattern (right of figure h1) is also based on layer-lines, which reflect the periodic repeat of the fibrous molecule. The intensity along the layer-lines is continuous and can be calculated via a "Fourier-Bessel Transform" of the repeating structure of the fibrous molecule (the Fourier-Bessel Transform replaces the Fourier transform used in standard crystallography - The Fourier-Bessel transform arises because of the cylindrical symmetry).
Diffraction from a helix
Calculating the x-ray diffraction pattern from a helix was of central significance in the development of molecular biology. It was first described by Francis Crick in his doctoral thesis. He wished to understand the diffraction to be expected from an [alpha]-helix. However, the theory was very quickly applied to determining the structure of DNA.

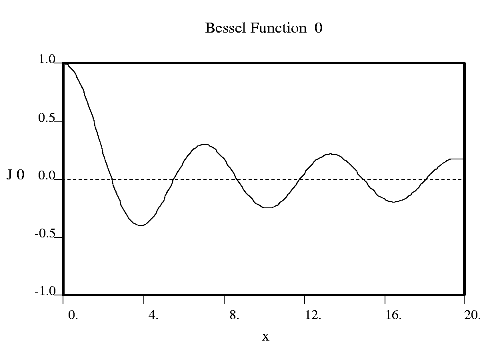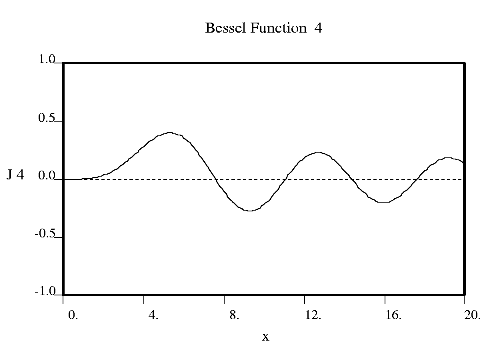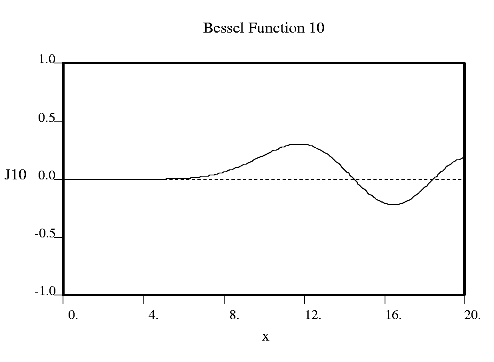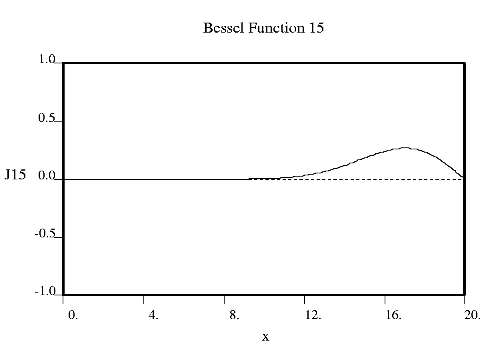
© Kenneth C. Holmes
Fourier used Bessel functions to calculate the flow of heat in cylindrical objects. Bessel functions characteristically begin with a strong peak and then oscillate like a damped sine wave as x increases. The position of the first strong peak depends on the order n of the Bessel function. A Bessel function of order zero begins in the middle of the pattern, a Bessel function of order 5 has its first peak at about x = 7, a Bessel function of order 10 does everything roughly twice as far out.
© Kenneth C. Holmes
Crick showed that the diffraction from a helix occurs along a series of equidistant lines rather than the Bragg spots one obtains from a three dimensional crystal. These lines (known as layer-lines) are at right angles to the axis of the fiber and the scattering along each layer-line is made up from Bessel functions. In helical diffraction Bessel functions take the place of sines and cosines one uses for crystals: Bessel functions (written Jn(x), where n is called the order and x the argument) are the form that waves take in situations of cylindrical symmetry (e.g. the waves you get if you throw a pebble into the middle of a pond). Bessel was a German astronomer who calculated accurately the orbits of the planets. Fourier used Bessel functions to calculate the flow of heat in cylindrical objects. Bessel functions characteristically begin with a strong peak and then oscillate like a damped sine wave as x increases. The position of the first strong peak depends on the order n of the Bessel function. A Bessel function of order zero begins in the middle of the pattern, a Bessel function of order 5 has its first peak at about x = 7, a Bessel function of order 10 does everything roughly twice as far out.
. In Fig h3 we show a continous helix and its diffraction pattern. Because the order of Bessel function increases with layer line number so does the position of the first strong peak. which then form the characteristic \"helix cross\". The position of the first strong peak is also inversely proportional to the radius of the helix. The spacing of the layer-lines is reciprocal to the pitch (P) of the helix. There is a reciprocal relationship between the layer line separation and the pitch- small separation large P, large separation small P.")

© Kenneth C. Holmes
Fig h3: A continuous helix and its diffraction pattern
© Kenneth C. Holmes
Crick showed that for a continuous helix the order of Bessel function n occuring on a certain layer line is the same as the layer line number l (counted from the middle of the diffraction pattern). In Fig h3 we show a continous helix and its diffraction pattern. Because the order of Bessel function increases with layer line number so does the position of the first strong peak. which then form the characteristic "helix cross". The position of the first strong peak is also inversely proportional to the radius of the helix. The spacing of the layer-lines is reciprocal to the pitch (P) of the helix. There is a reciprocal relationship between the layer line separation and the pitch- small separation large P, large separation small P.
 and by what angle you turn ([phi]) between one subunit and the next. This is enough to define a helix. Directly derivable from these parameters is the pitch P. The main effect of shifting from a continous to a discontinuous helix is to introduce new helix crosses with their origins diplaced up and down the axis (meridian) of the diffraction pattern by a distance 1/p. The diffraction pattern of a discontinous helix (with 10 subunits in one turn) is shown in Fig h4. Note that the layer-lines can be grouped into two kinds: those which are strong on the meridian of the fiber diffraction pattern (meridional) and those which have no intensity on the meridian (non-meridional). For a simple helix which repeats in one turn the fundamental layer line repeat is 1/P. The distance out along the meridian of the first meridional layer-line (not counting the origin) gives 1/p.")

© Kenneth C. Holmes
Fig h4: A discontinuous helix and its diffraction pattern
© Kenneth C. Holmes
However, real helicies are not continuous, rather they consist of repeating groups of atoms or molecules.The symmetry of a discontinuous helix can be defined in a number of ways: the most general is to quote how far one goes along the axis from one repeating subunit to the next in the macromolecule (the rise per residue p) and by what angle you turn (Π) between one subunit and the next. This is enough to define a helix. Directly derivable from these parameters is the pitch P. The main effect of shifting from a continous to a discontinuous helix is to introduce new helix crosses with their origins diplaced up and down the axis (meridian) of the diffraction pattern by a distance 1/p. The diffraction pattern of a discontinous helix (with 10 subunits in one turn) is shown in Fig h4. Note that the layer-lines can be grouped into two kinds: those which are strong on the meridian of the fiber diffraction pattern (meridional) and those which have no intensity on the meridian (non-meridional). For a simple helix which repeats in one turn the fundamental layer line repeat is 1/P. The distance out along the meridian of the first meridional layer-line (not counting the origin) gives 1/p.
Helix Selection Rule
For more complicated helices which repeat after two or more turns n and l are related by the helix selection rule
l = tn + um
The selection rule is an integer equation which makes use of an alternative definition of helix symmetry: there are t subunits in u turns of a repeat. m can take all positive and negative integer values. As an example, for an [alpha] helix t=5 and u=18 i,e, there are 18 subunits arranged on 5 turns per repeat. Solutions to the selection rule tell you which Bessel functions will turn up on which layer lines. Bessel functions with very large orders can be forgotten since they will occur so far out in the diffraction pattern (at such high resolution) that they will not be visible. Converseley, if one can figure out which Bessel functions turn up on which layer lines one knows the symmetry. The effects helical symmetry are in fact very useful for an analysis of fiber diffraction patterns. Without helical symmetry all Bessel functions would turn up on all layer lines, which would be a mess. The helical symmetry limits the allowed Bessel to one or two per layer-line which renders such problems tractable.
How this applies to DNA
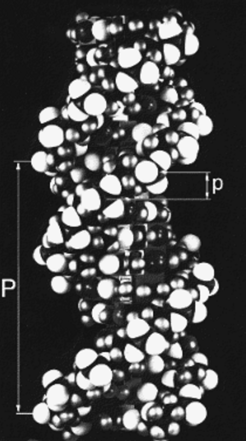
 and the pitch is ten times greater (i.e. P = 34Å. For low order layer lines the order of the Bessel function n which occurs on the l'th layer line is l. Because of their mass the phosphate groups are the dominant scatterers in a nucleic acids.The phosphate oxygens show up as prominent white spheres in the atomic model (Fig h5). The spacing of the layer lines in Fig h1 corresponds to 34Å - the helix repeats in 34Å. In this case this is also the pitch P. Knowing this and Francis Crick's formula for the scattering of a helix, Jim Watson was able to glean the radius of the phosphate groups from the look of the helix cross in Rosalind Franklin's fiber diffraction patterns of DNA . Furthermore, the strong meridional reflexion (see Fig h1) which has a Bragg spacing of 3.4Å, must correspond to 1/p, (i.e. the spacing between bases was 3.4Å). These pieces of information went a long way towards defining the essential parameters of the Watson-Crick model.")
© Kenneth C. Holmes
Fig h5: A space filling model of DNA-B form (W. Fuller)
© Kenneth C. Holmes
DNA-B form is a simple helix which repeats in one turn. It has 10 base pairs per turn so that the angle turned ber base Π is 36°. The spacing between the bases is 3.4 Å (i.e. p = 3.4 Å) and the pitch is ten times greater (i.e. P = 34 Å. For low order layer lines the order of the Bessel function n which occurs on the l'th layer line is l. Because of their mass the phosphate groups are the dominant scatterers in a nucleic acids.The phosphate oxygens show up as prominent white spheres in the atomic model (Fig h5). The spacing of the layer lines in Fig h1 corresponds to 34 Å - the helix repeats in 34 Å. In this case this is also the pitch P. Knowing this and Francis Crick's formula for the scattering of a helix, Jim Watson was able to glean the radius of the phosphate groups from the look of the helix cross in Rosalind Franklin's fiber diffraction patterns of DNA . Furthermore, the strong meridional reflexion (see Fig h1) which has a Bragg spacing of 3.4 Å, must correspond to 1/p, (i.e. the spacing between bases was 3.4 Å). These pieces of information went a long way towards defining the essential parameters of the Watson-Crick model.